
3D“硬度开关”如何重塑EVs并加速肿瘤进展?——基于TOCNF/GelMA纳米纤维水凝胶的仿生研究解读
摘要
肿瘤微环境中,细胞外基质(ECM)刚度与细胞外囊泡(EVs)均被认为是推动肿瘤进展的重要因素,但关键问题在于:ECM刚度升高,会不会把EVs也“重新编程”——不仅改变它的物理特性,还改变它携带的蛋白或miRNA,从而更强力地促进肿瘤表型增强?尽管已有研究在二维(2D)培养体系中揭示了ECM刚度对EVs的调控作用,但2D很难复刻真实肿瘤组织的三维结构与力学约束。基于此,本研究围绕”肿瘤微环境硬化是否通过细胞外囊泡(extracellular vesicles, EVs)放大促肿瘤信号”这一问题,构建了一个结构仿ECM且刚度可控的3D纳米纤维水凝胶平台(TOCNF/GelMA),并以“材料可控性→生物安全性→EV物性与负载差异→功能验证→机制必要性验证”的顺序完成证据链。

一、研究问题与核心假设:在3D ECM 样环境中硬化ECM是否通过EV放大促肿瘤信号?
1)临床与病理学背景:多种实体瘤伴随ECM沉积与纤维化,组织硬度上升常与侵袭性增强相关。
2)科学问题:在仿生 3D 肿瘤样 ECM 中,基质刚度是否会重塑肿瘤细胞分泌的 EV 的理化性质与负载(蛋白/miRNA),并通过激活受体细胞 MAPK/ERK1/2 通路来增强促肿瘤表型?
3)方法学空白:既往大量证据来自2D刚度基底,难以复现3D纤维网络的空间约束、受力方式与细胞-基质互作,导致外推性存疑。
据此,本研究提出假设:在3D ECM样环境中,基质硬度可重塑EV货物并增强其促肿瘤效应;受体细胞的MAPK/ERK轴可能为关键下游通路。
二、研究设计总览:从变量控制到机制必要性检验的证据链
研究设计遵循“材料可控性→生物安全性→EV物性与负载差异→功能验证→机制必要性验证”的路径:
1)材料学层面:构建结构仿ECM的3D纳米纤维水凝胶平台,并在尽量保持孔结构/纤维尺度等微结构可比的前提下仅分档调控刚度(软/硬),以确保“硬度”成为主要自变量,奠定后续差异可归因的实验基础,然而,该平台在生物学层面的可用性与生物安全性(如细胞长期存活、应激反应等)仍需进一步验证;
2)生物安全性:在两种刚度条件下验证细胞可长期存活并形成稳定3D肿瘤球,以排除材料毒性或培养应激导致的EV偏移,并保证EV来源稳定、可重复、组间可比;
3)EV层面:在细胞状态可比的前提下系统比较SoEVs与StEVs的载体物性(粒径/形态、电性、力学等)及负载谱(蛋白/miRNA等),从“EV本体是否被重塑”这一关键前置问题出发,确认硬度对EV输出具有系统性影响;
4)功能层面:进一步将“EV差异”转化为“生物学后果”,以体外迁移/增殖及体内移植瘤生长为终点,检验不同硬度来源EV是否产生可观察的促肿瘤表型,从而把研究从描述性差异推进到病理学意义验证;
5)机制层面:在明确功能差异后,利用组学富集筛选关键通路,并通过受体细胞通路磷酸化确认信号激活,再以药理学抑制进行干预检验其对表型的影响,从而完成“相关→必要性”的因果验证闭环。
三、主要结果:逐步回答“材料可控性→生物安全性→EV物性与负载差异→功能验证→机制必要性验证”的关键问题
3.1 模型搭建:TOCNF/GelMA 3D纳米纤维ECM如何实现“软/硬切换”?
1)材料与平台:TOCNF(TEMPO氧化纤维素纳米纤维)+ GelMA(甲基丙烯酰化明胶)在405 nm光交联下形成稳定3D水凝胶网络。
TOCNF提供类胶原的纳米纤维网络,表面羧基丰富,更接近天然ECM的纤维结构特征;GelMA提供RGD黏附位点,支持细胞黏附、伸展与功能表达
2)结构与力学:不仅“像ECM”,还“可控”
SEM及统计显示孔径主要集中在20–30 μm,形成更贴近真实组织的三维微环境;同时可通过“软/硬”两类水凝胶实现不同刚度调控。
该体系的核心目的在于同时满足“3D纤维结构仿生”与“细胞可黏附、可长期培养”两项关键前提,从而为后续研究提供一个“结构可比、刚度可控”的3D微环境,使EV差异分析更有可能被可靠地归因于基质硬度,而非由微结构差异或黏附不足引发的非特异性效应所驱动;然而,该平台是否具备足够的生物安全性与生物学可用性(如长期细胞存活)仍需进一步验证。
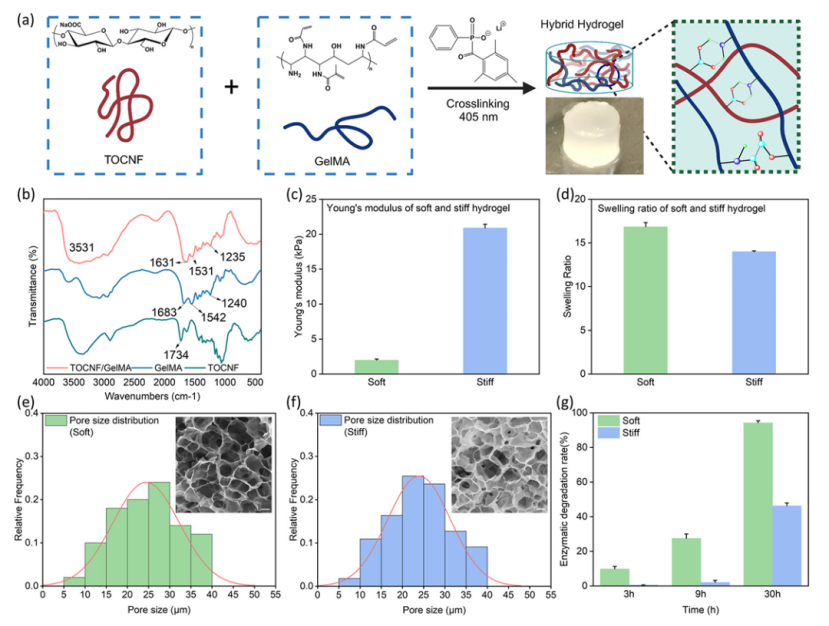
图1:TOCNF/GelMA复合水凝胶的制备及表征
3.2生物相容性:平台稳定支持3D生长,为EV差异分析奠基
在两种刚度条件下,细胞在连续15天的3D培养中仍维持较高存活率(>80%),表明该水凝胶平台具备支持长期3D培养与持续EV释放的生物学可用性及生物安全性,从而在进入EV物性与货物差异比较前,有效降低了“材料毒性/培养应激导致EV偏移”的潜在影响,为后续将EV差异更可靠地归因于基质刚度提供了更稳健的生物学基础,并且,在此“细胞状态可比、EV来源可靠”的前提下,下一步关键在于回答:基质硬度是否改变EV本体的载体属性。
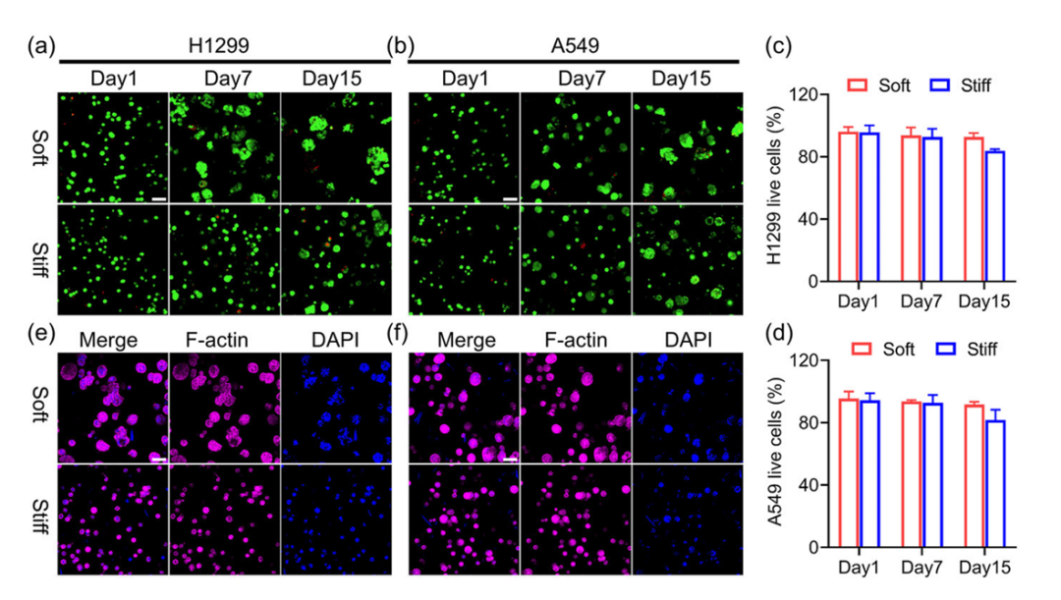
图2:生物相容性测试
3.3 关键前置验证:SoEVs与StEVs的粒径控制与膜属性差异确认
SoEVs与StEVs在形态上均呈典型杯状/球形,粒径分布相近且主要集中于100–200 nm,说明两组EV在“尺寸维度”具备可比性;StEVs的zata电位绝对值减小(表面负电减少)并伴随力学性质下降(Young’s modulus更低、更“软”),表明基质硬度可能重塑了EV膜的物理化学状态。
在此“尺寸可比”的前提下,后续开展蛋白/miRNA货物谱差异分析与功能验证更不易被摄取偏差所解释,并为建立“硬度→EV载体属性/膜状态改变→货物与信号传递差异→生物学效应差异”的证据链提供了合理起点,并进一步提出下一层关键问题:不同硬度来源EV是否会转化为可量化的促肿瘤表型,并在体外与体内环境中保持一致?
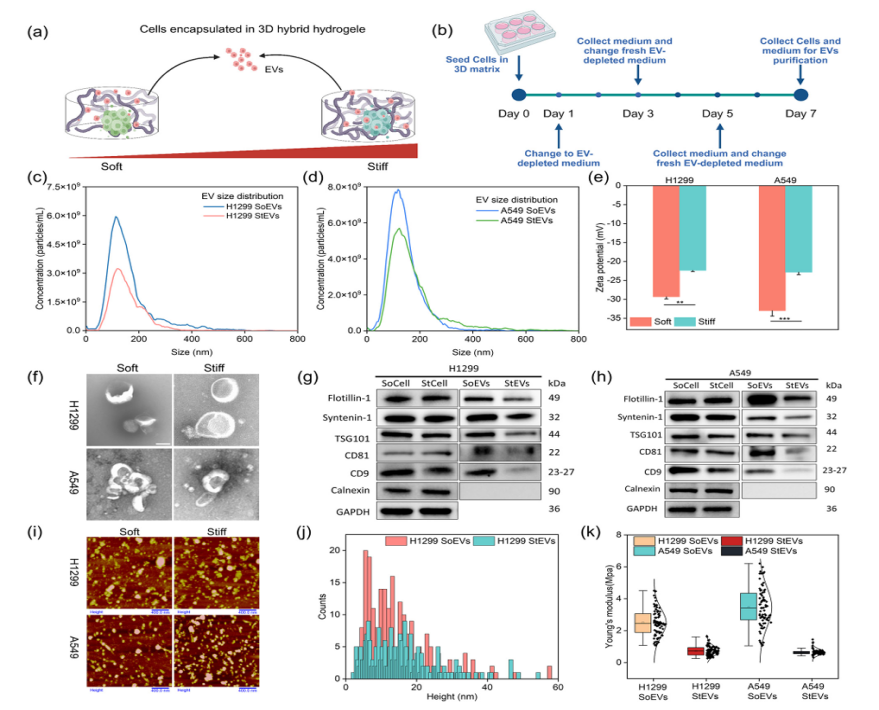
图3:3D培养以及EV分离与表征
3.4 功能层面:StEVs增强迁移/增殖并促进体内肿瘤生长
尽管前期已发现不同硬度来源EV在物性与货物谱上存在差异,但为将其从“描述性发现”推进到具有病理学意义的功能证据,作者在体外/体内模型中对其促肿瘤效应进行验证。
1) 体外层面:迁移与增值增强
StEVs相较SoEVs/PBS更明显促进肿瘤细胞迁移及肿瘤细胞增殖率,并呈现更强的促肿瘤表型。
2)体内层面:移植瘤模型验证“促瘤效应”
经尾静脉给予StEVs的小鼠在5周内肿瘤体积增长速率提高2.1倍,终末瘤重增加约1.8倍,生物发光信号强度显著高于SoEVs与PBS组,提示StEVs通过系统性循环显著加速了肿瘤的生长与进展。
体外—体内一致的功能证据回答了“不同硬度来源EV的差异是否具有可观察的病理学后果”,但这一证据仍停留在“效应确认”,尚不足以解释其分子基础。因此,研究的下一步关键问题为:StEVs究竟通过哪些负载变化驱动受体细胞的迁移/增殖增强,并可能激活哪些下游信号通路?
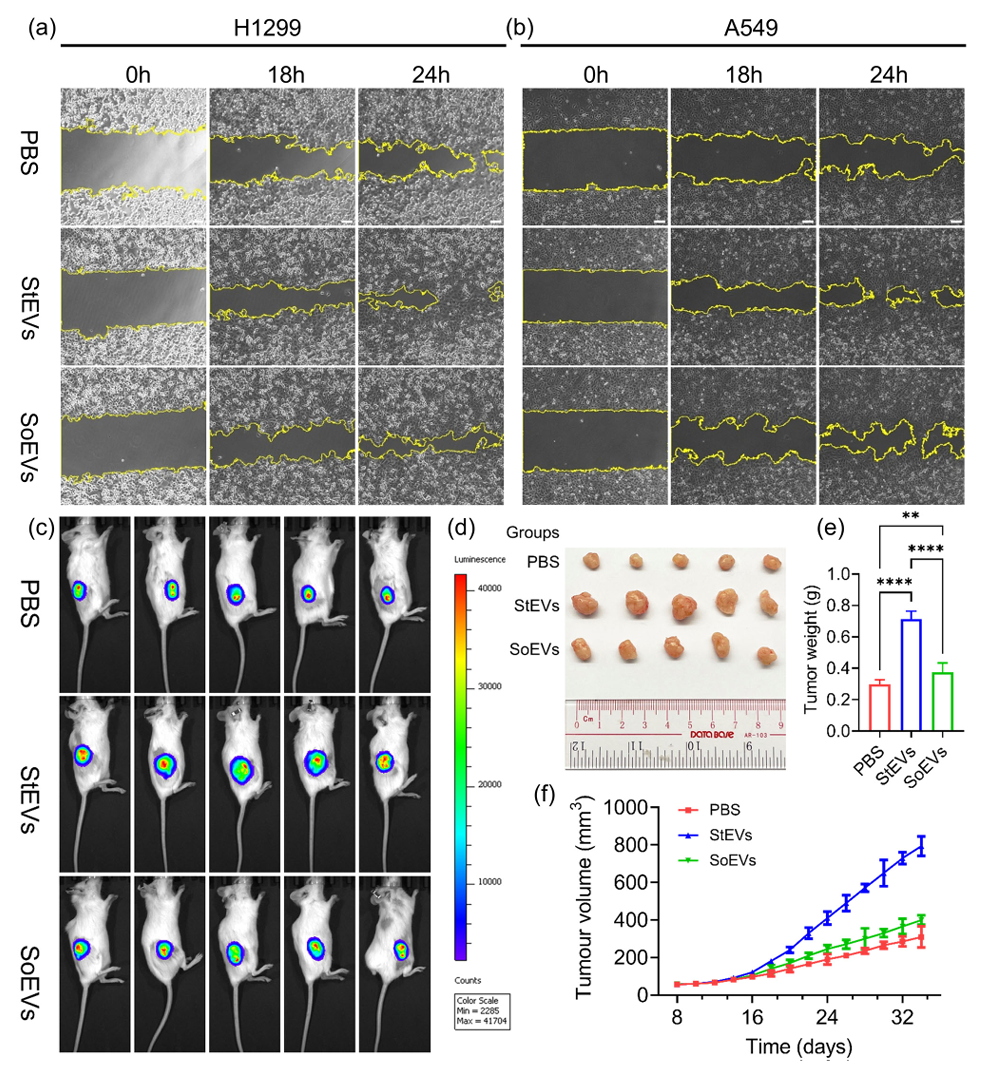
图4:StEVs促进迁移与体内肿瘤生长(划痕、IVIS、终点肿瘤重量、体积曲线)
3.5机制路径搭建:蛋白/miRNA组学与通路富集指向MAPK/ERK轴
前一阶段已证实StEVs在体外与体内均表现出更强的促肿瘤效应,研究亟需从“现象”进一步推进到“机制”:即明确哪些EV货物变化在驱动该表型、并通过哪条信号通路实现。
1)组学证据提示:硬基质相关EV蛋白货物改变并指向MAPK等促癌通路
差异蛋白量级很大:SoEVs与StEVs共享蛋白3965个,但StEVs相对SoEVs上调502、下调165个蛋白,说明硬基质在“重写EVs货物清单”。
通路富集直指促癌网络:KEGG富集出现MAPK、PI3K-Akt、ECM-receptor interaction等与迁移、增殖、微环境互作高度相关通路。
基于蛋白组学的无偏分析,硬基质相关EVs的蛋白货物发生系统性改变,并在通路层面提示MAPK等候选机制,但是尽管蛋白组学与KEGG富集为机制提供了方向性线索,但富集结果本质上属于“候选通路层面的关联提示”,并不能替代受体细胞内真实信号激活的证据,要确定其在受体细胞中是否真正被激活,需直接检测下游信号读出。
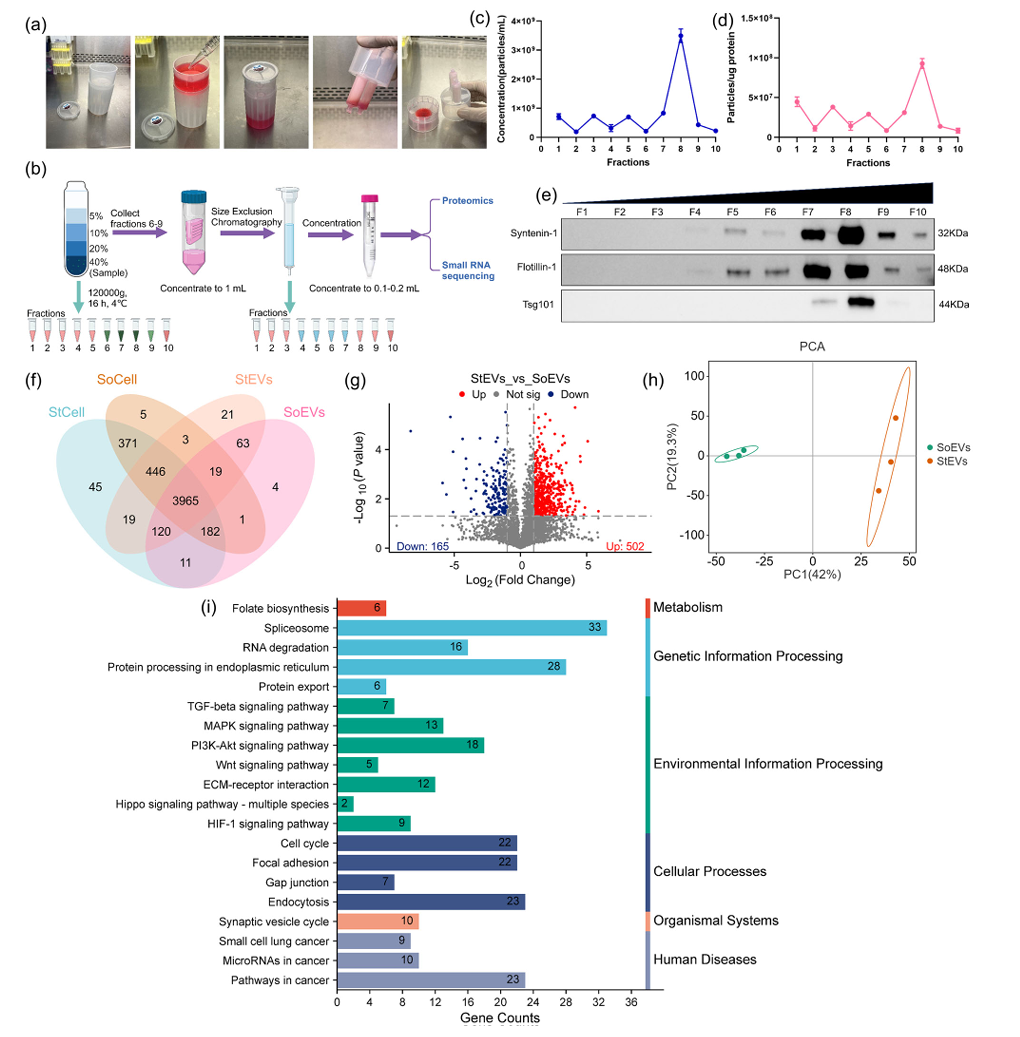
图5:EV蛋白组学(两步纯化、火山图、PCA、通路富集)
2)通路层面验证:StEVs在受体细胞中以ERK1/2磷酸化为主要响应
为进一步从“候选”走向“被激活的通路”,作者在受体细胞中检测关键通路的磷酸化水平以作为功能性读出,以判定哪些信号轴在StEVs刺激下被优先点亮并可能介导迁移/增殖表型。
ERK1/2磷酸化最突出:StEVs组p-ERK1/2显著升高,明显强于SoEVs与PBS。
其他通路相对弱:PI3K-Akt、TGF-β、JAK-STAT等虽可能参与,但激活程度不如ERK突出。
上述结果为后续实验提供三方面支撑:一是将机制验证从“盲试”转为“有据可依的聚焦”,优先锁定MAPK/ERK通路;二是提供可量化的关键机制读出(p-ERK1/2),便于评估干预是否真正切断信号传递;三是为因果检验建立清晰路径,即在“StEVs→ERK激活→迁移/增殖增强”的框架下,进一步通过ERK抑制剂(如U0126)验证该通路对表型的必要性,从而把证据由相关性推进到因果支持。
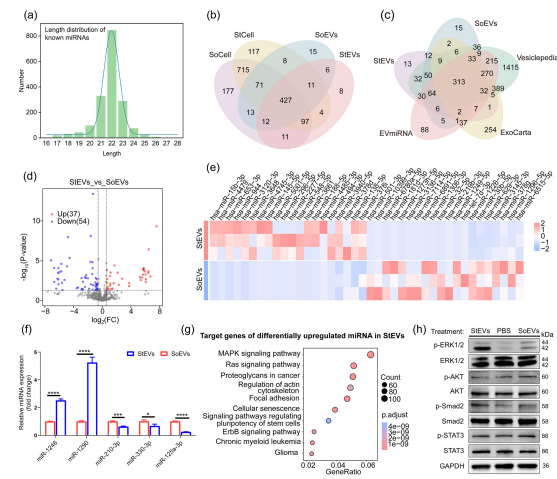
图6:EV小RNA测序与通路预测(差异miRNA、qPCR验证、KEGG富集)
3)必要性检验:ERK抑制削弱表型,支持“EV→ERK→促肿瘤”链条
引入ERK通路抑制剂U0126进行干预后:
迁移被逆转:StEVs促进迁移,但在U0126作用下显著受抑。增殖也被按停,并且通路验证闭合:StEVs促进增殖,U0126显著削弱。WB进一步证明总ERK基本不变,而p-ERK被U0126拉回。
该结果直接回答了ERK轴是否为StEVs促肿瘤表型的重要依赖环节,并将证据从“相关性提示”推进到“必要性支持”。
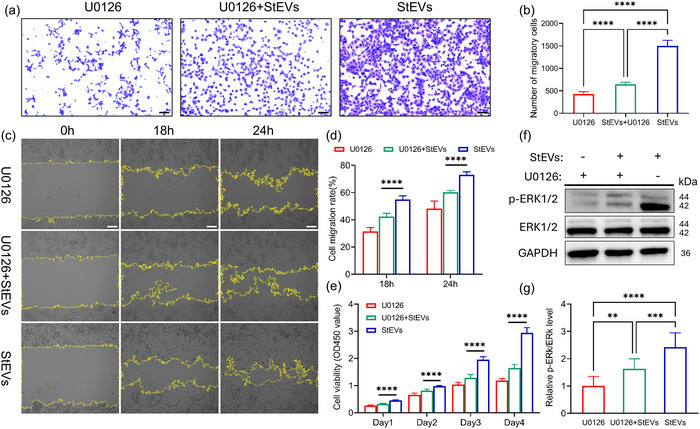
图7:U0126证明StEVs促迁移/促增殖依赖MAPK/ERK1/2
3)完整调控机制:
硬基质→EV cargo重塑→受体细胞ERK1/2优先激活→迁移/增殖增强;U0126可逆转表型并降低p-ERK,证明StEVs促肿瘤效应对MAPK/ERK1/2具有依赖性。
四、创新点剖析
1)将ECM力学刚度从“背景特征”提升为可实验操控的上游调控变量,用于解析EV介导的通讯机制。
2)以3D仿生纳米纤维基质替代传统2D刚性培养底板,提高结论的生理相关性与可外推性。
3)TOCNF提供类胶原纳米纤维支架 + GelMA提供RGD黏附位点,在结构与细胞相互作用层面更接近天然ECM。
4)多模态表征将EVs视作“物理对象”进行定量研究(电性、力学性质等),补充了传统“粒径/标志物”范式的不足。
总体归纳:研究构建了TOCNF/GelMA仿生3D纳米纤维ECM平台,在较好控制混杂因素的前提下系统比较不同刚度来源EVs的理化特性与功能,并结合蛋白/miRNA组学、通路验证与抑制剂因果检验,建立了“ECM刚度—EV cargo重塑—ERK依赖的促肿瘤表型”的证据链闭环
五、局限性及展望
1)局限性
1.缺乏患者样本与临床相关性验证,尚难建立“硬度—EV特征—临床结局”的关联链。
2. 动物模型更偏“证明生物学可发生”,与临床原位肿瘤、免疫系统与人群异质性仍存在差距。
3. “可干预性”目前主要停留在体外药理学验证层面,仍需更贴近临床的体内验证与分层策略。
3)展望
下一步值得从差异cargo中锁定关键驱动分子(而非仅停留在通路层面),并构建多级刚度梯度获得剂量—反应规律;同时将模型拓展至CAF/免疫细胞共培养与患者来源体系以验证普适性与临床相关性。结合脂质组学/表面蛋白组等多组学,有望补齐“膜组成—物理性质—摄取—功能”的机制闭环,并为“力学微环境—EV通讯轴”的干预策略提供更明确的靶点线索。